Inheritance: Autosomal dominant (Romano-Ward syndrome, penetrance 25–75% for QTc >460ms); rare autosomal recessive (Jervell-Lange-Nielsen syndrome with congenital deafness, homozygous/compound heterozygous KCNQ1 or KCNE1 variants, nearly 100% penetrance).
Genetic yield: ~75% with comprehensive testing. Up to 25% are gene-elusive, but phenotypic severity mirrors genotype-positive LQTS in these patients (Asatryan et al 2024, Circ Arrhythm Electrophysiol)[8]
ClinGen-validated genes:
| Gene | Subtype | % Genotype+ | Channel / Mechanism | Arrhythmic Triggers |
|---|
| KCNQ1 | LQT1 | ~50% | IKs loss-of-function (K⁺) | Exercise, swimming, adrenergic-dependent |
| KCNH2 | LQT2 | 35–40% | IKr loss-of-function (K⁺) | Auditory triggers, emotion, postpartum; higher risk in females |
| SCN5A | LQT3 | ~10% | INa gain-of-function (Na⁺ delayed inactivation) | Rest/sleep, bradycardia; mexiletine genotype-specific benefit |
| CALM1/2/3 | Calmodulinopathy | <1% | Calmodulin (impaired Ca²⁺-channel inactivation) | Often de novo; extreme QTc; severe early-onset, high arrhythmic risk; neurodevelopmental features |
| TRDN | – | <1% | Triadin; autosomal recessive | Exercise-triggered; T-wave abnormalities at rest |
| CACNA1C | LQT8 (Timothy) | Rare | ICaL gain-of-function (Ca²⁺) | Multiorgan syndrome: ASD, autism, syndactyly; very rare |
Gene-elusive LQTS (~11–25%): Multiple mechanisms, polygenic risk score contribution, environmental QTc modulators, exercise-induced repolarisation abnormalities, new causal genes not yet identified, or variant reclassification over time. Management mirrors genotype-positive LQTS. Referral to specialised cardiogenetic clinic recommended (Asatryan et al 2024)[8].
Modifier genes: Common variants modulate QTc and arrhythmic risk in individual patients, polygenic risk scores can refine risk stratification beyond the primary pathogenic variant (Schwartz & Crotti, NEJM 2025).
Genotype-Specific Management (Zhu et al 2024, Schwartz & Crotti NEJM 2025)
[7][2][9]
| Subtype | Gene | % LQTS | Triggers | ECG Pattern | Key Management Points |
|---|
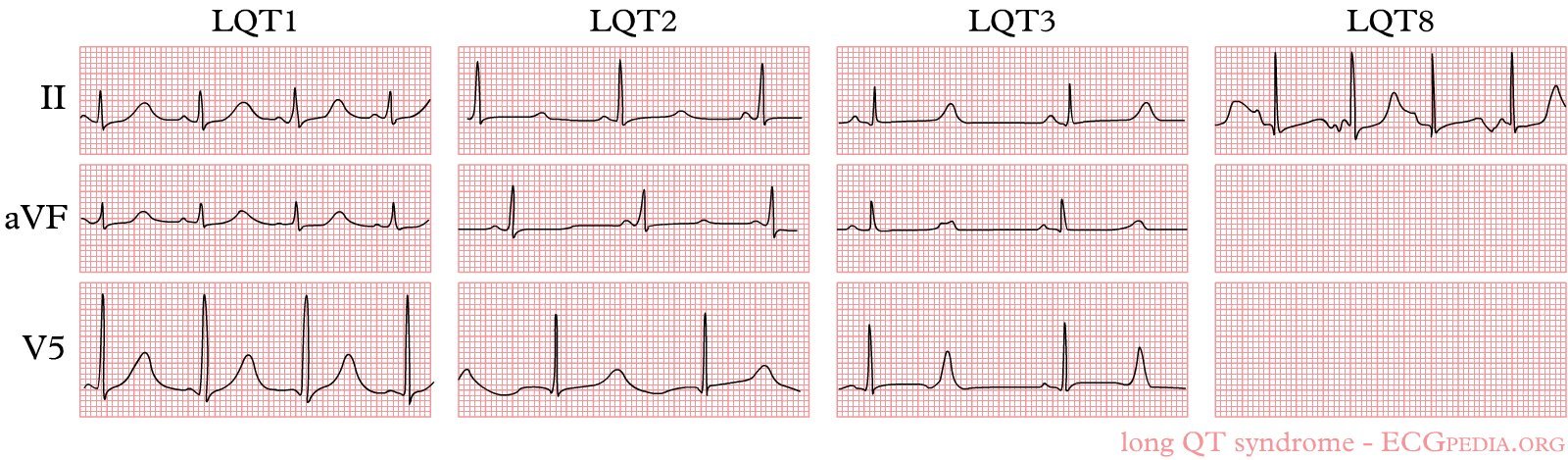
| LQT1 |
KCNQ1 |
~50% |
Exercise, swimming (adrenergic) |
Broad-based T wave |
Beta-blockers most effective; nadolol preferred; restrict competitive swimming |
| LQT2 |
KCNH2 |
35–40% |
Auditory triggers, emotion, postpartum |
Notched/bifid T wave |
Higher risk in females; avoid alarm clocks; K⁺ supplementation; beta-blockers effective |
| LQT3 |
SCN5A |
~10% |
Rest/sleep, bradycardia |
Long isoelectric ST segment |
Mexiletine (off-label): shortens QTc, reduces events; beta-blockers less effective; pacemaker in selected cases |
| Calmodulinopathies |
CALM1/2/3 |
<1% |
Variable; often de novo |
Extreme QTc prolongation |
Very high risk; early ICD; consider LCSD; flecainide may have role; specialist centre |